|
One of the first molecular changes in the Ube3a m-/p+ mouse to be identified was an alteration in phosphorylation levels of the calcium/calmodulin-dependent kinase type 2 (CaMKII). Analysis showed an increase in phosphorylation at the autophosphorylation sites Threonine 286 (Thr286) and Threonine 305 (Thr305). This corresponded with a significant reduction in calcium-calmodulin sensitivity, reduced basal CaMKII activity and a decrease in total synaptic density-associated CaMKII (31). This observation was of particular interest in light of the large body of research demonstrating that the disruption of CaMKII function has profound effects on synaptic plasticity and long-term memory formation (32, 33). Specifically, site directed mutation at either Thr286 or Thr305 with an aspartate, which mimics persistent phosphorylation, produces devastating effects on memory formation (33-35).
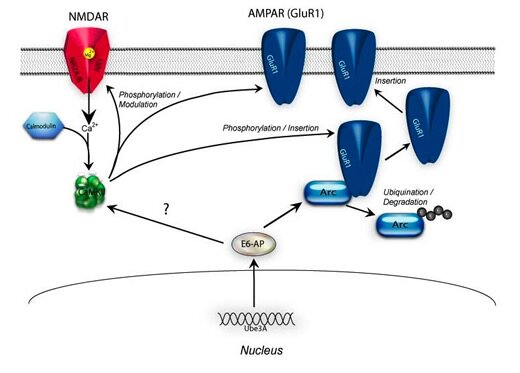
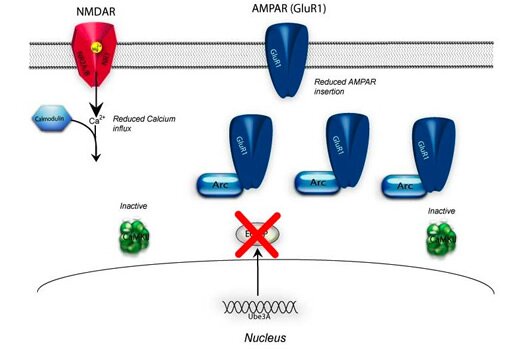
Figure (BELOW) Wildtype versus Angelman Syndrome synaptic biochemistry
In the normal synapse maternal Ube3a protein targets other proteins, such as Arc, for degradation. The causal relationship between Ube3a and CaMKII is unknown; however, the presence of Ube3a maintains phosphatase activity and CaMKII is held in a state and receptive to calcium influx following synaptic activation. In Angelman syndrome there is an increase in Arc protein that in-turn decreases AMAPR insertion into the membrane. In addition, CaMKII is found to be hyper-phosphorylated at the aoutoinhibitory site reducing basal CaMKII activity and reducing calcium-calmodulin-induced activation.
The Biochemistry in a Wildtype Mouse
The Biochemistry in an AS Mouse
Identification of the biochemical and functional alterations in CaMKII in the mouse model provoked the obvious question; To what extent are the Ube3a m-/p+ phenotypes due to the misphosphorylation of CaMKII? Because of the severe detrimental effects of CaMKII Thr305 phosphorylation on CamKII enzymatic activity, as well as the similarities in the phenotypes of the CaMKII Thr305 to Asp305 mutant mouse with the AS mouse, the Thr305 site was hypothesized to be responsible for the major phenotypes seen in the Ube3a m-/p+ model.
Studies in the laboratories of Weeber and Elgersma utilized female AS mice crossed with heterozygous aCaMKII males that carried the targeted aCaMKII-Thr305 to Val305 mutation. The amino acid valine cannot be phosphorylated and thus prevents the accumulation of phosphorylated Thr305CaMKII (inactive CamKII) in the neurons. Importatnly, utilizing heterozygotes with this point mutation reduced but did not completely obliterate the inhibitory phosphorylation. Remarkably, mice with the dual CaMKII and Ube3A mutations showed a complete rescue of all the major phenotypes exhibited by AS mice. These studies confirmed the contribution of altered CaMKII to the phenotypes of the Ube3a maternal deficient mouse model and suggested that CaMKII dysfunction may serve as an underlying molecular mechanism of human AS.
This work was reported and is available to view in Nature Neuroscience.
Current NLML AS Research Projects
Introduction to Angelman Syndrome
UBE3A Gene Function
Model Systems of Angelman Syndrome
Parents Area
31. E. J. Weeber et al., J Neurosci 23, 2634 (Apr 1, 2003).
32. A. J. Silva, R. Paylor, J. M. Wehner, S. Tonegawa, Science 257, 206 (Jul 10, 1992).
33. M. E. Bach, R. D. Hawkins, M. Osman, et al., Cell 81, 905 (Jun 16, 1995).
34. K. P. Giese, N. B. Fedorov, R. K. Filipkowski, A. J. Silva, Science 279, 870 (Feb 6, 1998).
35. R. Bejar, R. Yasuda, H. Krugers, K. Hood, M. Mayford, J Neurosci 22, 5719 (Jul 1, 2002).
|
