|
THE AS MOUSE MODEL
Two distinct mouse models have been developed using different strategies to disrupt Ube3a. The first mouse line utilized an Epstein-Barr virus latent membrane protein 2A (Lmp2a) transgenic insertion to remove the entire equivalent AS critical region (4, 25). Alternatively, the mouse model developed by Jiang and colleagues utilized a specific maternal gene knockout in Ube3a via a null mutation (26). While both of these mouse models effectively disrupt maternal Ube3a expression, there are advantages and disadvantages for both models.
Multiple gene disruption in the deletion mouse model more closely resembles the genetic abnormalities seen in a majority of AS patients. However, multiple gene knockout models are inherently more complicated than single gene knockout models. In comparison, the deletion mouse model might not fully or accurately show the biochemical alterations leading to the true manifestation of AS due to the combined effect of several altered genes. Recently another mouse model has been developed through a large maternal deletion from the Ube3a gene to the Gabarb3 gene. This deletion is perhaps the most equivalent to the common deletion seen in human AS that removes both the Ube3a and Gabarb3 gene. Each of these models exhibits cognitive defects, motor coordination problems and increased seizure propensity.
Regardless of their respective caveats, the null mutation mouse model has become the standard for AS mouse model research and nearly all of the pertinent literature has used this specific mouse model.
Ube3a genetic imprinting
It should be noted that the Ube3a maternal deficient (m-/p+) mouse is also valuable for studying the specific patterns and mechanisms of maternal Ube3a imprinting in the CNS of developing and adult animals. This becomes especially important in light of the scarcity of AS human post-mortem brain tissue. The identification of imprinted regions through the use of immunohistochemical techniques in mice is commonly performed, and relatively easy to interpret. Importantly, both AS patients and Ube3a deficient mice exhibit normal brain histopathology, suggesting that maternal Ube3a deficiency does not affect neuronal development or organization. In adult mice the detection of Ube3A mRNA revealed high expression levels in areas CA1 and CA3 of the hippocampus, basal ganglia, cerebellum and throughout the cerebral cortex. This pattern was compared to maternal deficient mice that showed no detectable hippocampal expression, significant reduced cerebellar expression with no detectable expression in the purkinje cell layer, and an overall reduced expression in the olfactory region associated with a lack of detectable expression in the mitral cell layer (25, 26). These in situ results established a dogma of regional paternal protein expression of Ube3a, whereby the imprinted mechanism of Ube3a specifically creates a situation of Ube3a absence in selective brain regions.
Recently, brains from Ube3a maternal deficient mice were examined by western blot analysis for Ube3a protein. In contrast to what had been concluded from the in situ data, there was a complete absence of detectable Ube3a protein in the hippocampus, cerebellum, prefrontal cortex, striatum and associative areas and the remaining cerebral cortex (27). These finding indicate that maternal deficiency of Ube3a remarkably results in the global absence of Ube3a protein. This experiment, performed more than a decade after the original expression pattern report, raises an interesting question: Why are there detectable paternal Ube3a mRNA patterns that are regionally distinct, but no detectable paternal Ube3a protein in those same brain regions? It is unclear if the paternal Ube3a mRNA serves a biochemical purpose in the neuron per se, or if it can be translated into functional protein. To date there is no in vivo evidence showing increased paternal protein production under any circumstances. Regardless, it may explain why maternal Ube3a deficiency leads to wide-spread and multi-domain symptomology in children with AS.
PHENOTYPES OF THE AS MOUSE MODEL
The initial characterization of the Ube3a maternal deficient mouse mutant (referred to from here on as Ube3a m-/p+) revealed striking similarities to the human phenotype (26). These phenotypic similarities are more easily discernable when sub-divided into categories of physical, physiologic and behavioral characteristics.
Physical similarities: Motor coordination deficiencies are a common feature of human AS. Motor function and coordination in the mouse model was assessed using hind-paw footprint analysis, the bar crossing test and the accelerating rotorod test. Each of these tests confirmed a distinct and significant motor deficit in the Ube3a m-/p+ mice, which mimics the tremor, ataxia and motor coordination difficulties described in human AS patients. In addition, maternal deficient mice show a distinct cerebellar defect in licking behavior during drinking (28). Seizure in AS patients is very prominent and found to affect > 90% of diagnosed AS patients. Likewise, seizures in Ube3a m-/p+ mice could easily be induced through audiogenic means by running an object vigorously over the metal grate lid of the mouse’s home cage or with white noise over 100 dB (26).
 Figure 1. showing the Accelerating Rotorod Test Figure 1. showing the Accelerating Rotorod Test
This is a test to evaluate both motor coordination and motor learning abitilty. The rod accelerates from 4-40 rpm over a 3 minute time period. Latency to fall off the rod during a 4 trail per day / 2 day training paradigm.
Physiologic phenotypes: Naturally, physical changes are more easily quantifiable and tend to be a more convincing argument for the recapitulation of a human disorder “mouse model”. However, there are several ways to measure the behavioral correlates to human cognitive ability in the mouse by assessing function at a synaptic level through electrophysiologic techniques. Assessment of hippocampal CA1 synaptic plasticity showed a severe defect in high frequency stimulation (hfs)-induced long-term potentiation (LTP) using a standard 2 train 100Hz stimulation protocol. Increasing the number of hfs trains can produce a potentiation that is substantially higher in magnitude and longer lasting. In order to test the possibility that the LTP deficits in Ube3a m-/p+ mutants were due to an increase in the threshold of LTP induction, potentiation was induced with a saturating amount of hfs (6 trains at 100Hz) designed to give the maximum degree of potentiation. This hfs protocol elicited LTP in the Ube3a m-/p+ mouse to the same amount seen in wild-type controls suggesting that long-lasting potentiaion in area CA1 was possible if a stimulation threshold was exceeded. These observations fit nicely into a model of Ube3a-dependent Arc dysfunction of AMPAR synaptic activation (Figures 2 and 3).
Cognitive similarities: Overall cognitive ability has been assessed by both associative learning and memory using fear conditioning and spatial learning and memory using the hidden platform water maze test. For associative learning using the fear conditioning learning paradigm an aversive stimulus (unconditioned stimulus, US), consisting of a mild foot shock, is paired usually twice with an auditory component (conditioned stimulus, CS). The animals are then tested 24 hours following training through the reintroduction of the mouse into the conditioning chamber or placing the mouse in a novel context while repeating the CS. Learning and memory is assessed by observing freezing behavior, which is a learned behavioral fear response to either the association to the CS or context. Although both contextual and cued fear-conditioned learning is dependent on the proper function of the amygdala, only the contextual learning is hippocampus-dependent. The Ube3a m-/p+ mutant mice showed normal associative learning to the CS, but had a severe contextual fear conditioning deficit. Alterations in spatial learning were not as straight forward. Using a spaced training protocol Ube3a m-/p+ mice show similar decreased latency to find the platform following subsequent days of training. However, probe tests show a significant decrease in platform crossings and time spent in the target quadrant. This suggests that Ube3a m-/p+ mice develop a search strategy that is not as efficient as wild-type mice.
 Figure 2 (LEFT) Hidden Platform Water Maze Test Figure 2 (LEFT) Hidden Platform Water Maze Test
Spatial learning has been assessed using the Hidden Platform water maze. This test utilizes spatial cues located outside of the pool where the mouse is trained to use the spatial cues to find the platform. The latency to find the platform is a indicator of learning ability. Memory retention can be determined with a probe test where the platform is removed and the amount of time spent in the target quadrant or the number of platform crossings is determined.
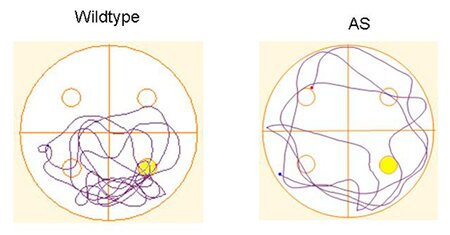
Figure 3 (BELOW)
Representative paths taken during a probe test for a wild type and angelman syndrome mouse model. The angelman syndrome mouse has a tendency to swim in large circles indicating a lack of an effective search strategy and a significantly decrease time in the target quadrant.
Current NLML AS Research Projects
Introduction to Angelman Syndrome
UBE3A Gene Function
Molecular Changes in the AS Mouse Model
Parents Area
4. J. M. Gabriel et al., Proc Natl Acad Sci U S A 96, 9258 (1999).
25. U. Albrecht et al., Nat Genet 17, 75 (1997).
26. Y. H. Jiang et al., Neuron 21, 799 (1998).
27. R. M. Gustin et al., Neurobiol Dis 39, 283 (Sep).
28. D. H. Heck, Y. Zhao, S. Roy, et al., Hum Mol Genet 17, 2181 (Jul 15, 2008).
|
